
Cała zawartość iLive jest sprawdzana medycznie lub sprawdzana pod względem faktycznym, aby zapewnić jak największą dokładność faktyczną.
Mamy ścisłe wytyczne dotyczące pozyskiwania i tylko linki do renomowanych serwisów medialnych, akademickich instytucji badawczych i, o ile to możliwe, recenzowanych badań medycznych. Zauważ, że liczby w nawiasach ([1], [2] itd.) Są linkami do tych badań, które można kliknąć.
Jeśli uważasz, że któraś z naszych treści jest niedokładna, nieaktualna lub w inny sposób wątpliwa, wybierz ją i naciśnij Ctrl + Enter.
Chłoniak limfoplazmocytowy B-komórkowy Waldenströma
Ekspert medyczny artykułu

Ostatnia recenzja: 12.07.2025
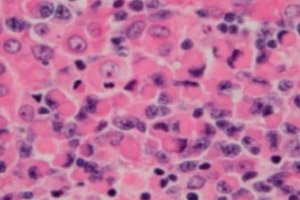
Złośliwe zaburzenie limfoproliferacyjne (immunoproliferacyjne), chłoniak limfoplazmocytowy lub makroglobulinemia Waldenströma to nowotwór komórkowy małych limfocytów B – komórek B, które zapewniają funkcje ochronne układu limfatycznego i odporność humoralną organizmu. Rozpoznanie powinno być postawione dopiero po wykluczeniu wszystkich innych małych chłoniaków B-komórkowych. Makroglobulinemia Waldenströma została opisana w 1944 roku przez Jana G. Waldenströma, który zgłosił nietypowe objawy krwawienia limfadenopatycznego, niedokrwistości, zwiększonej OB, hiperlepkości i hipergammaglobulinemii u dwóch pacjentów. [ 1 ], [ 2 ]
Epidemiologia
Ten typ chłoniaka jest rzadkim, wolno rozwijającym się nowotworem hematologicznym, a statystyki kliniczne szacują, że jego wykrywalność w tej grupie chorób wynosi około 2%. Co więcej, liczba chorych płci męskiej jest prawie dwukrotnie większa niż liczba kobiet.
Według niektórych danych zapadalność na chłoniaka limfoplazmocytowego w krajach europejskich wynosi 1 na 102 tysiące osób rocznie, a w USA – 1 na 260 tysięcy. [ 3 ]
Przyczyny chłoniak limfoplazmocytowy
Do tej pory etiologia większości chorób onkologicznych pozostaje nieznana, ale badania nad podłożem genetycznym niektórych z nich trwają. Badając przyczyny złośliwych chorób komórek plazmatycznych, w tym chłoniaka limfoplazmocytowego komórek B –makroglobulinemię Waldenströma, naukowcy odkryli związek między patologiczną proliferacją (podziałem komórek) limfocytów B na późnym etapie ich różnicowania a obecnością pewnych zaburzeń molekularno-genowych, które zmieniają podstawowe funkcje komórkowe.
U chorych na makroglobulinemię Waldenströma zidentyfikowano zmiany w niektórych genach – mutacje somatyczne, czyli takie, które dotyczą tylko tkanek, w których doszło do uszkodzenia genów odrębnej populacji klonalnej komórek i tworzą warianty ich genomu, co prowadzi do zaburzeń cyklicznych i strukturalnych na poziomie komórkowym.
Przede wszystkim są to mutacje somatyczne genu MYD88 (L265P) i CXCR4, kodującego białko cytozolowe, które jest ważne dla wrodzonej i adaptacyjnej odpowiedzi immunologicznej: jako adapter zapewnia transmisję sygnałów od mediatora prozapalnego IL-1 (interleukiny-1) i komórek receptora typu Toll, które aktywują odpowiedź immunologiczną. W wyniku mutacji somatycznej powstają anomalie w łańcuchu polipeptydowym cząsteczki tego białka – jego podstawie strukturalnej. [ 4 ]
Czynniki ryzyka
Oprócz ogólnych czynników ryzyka (narażenie na wysokie poziomy promieniowania, rakotwórcze substancje chemiczne itp.), poniższe czynniki uważa się za predyktory zwiększonego prawdopodobieństwa wystąpienia makroglobulinemii Waldenströma jako choroby limfoproliferacyjnej niskiego stopnia:
- podeszły wiek (powyżej 65 lat);
- obecność krewnych z tą diagnozą, a także z chłoniakiem nieziarniczym B-komórkowym lub przewlekłą białaczką limfocytową;
- przewlekłe zapalenie wątroby typu C;
- historia łagodnej gammapatii monoklonalnej, idiopatycznej choroby hematologicznej, której istotą jest produkcja nieprawidłowo zmienionych gamma-globulin typu M przez komórki plazmatyczne limfocytów;
- choroby autoimmunologiczne, w szczególności zespół Sjogrena.
Patogeneza
W wyniku kontaktu z antygenem lub stymulacji przez limfocyty T, część limfocytów B przekształca się w komórki plazmatyczne – limfocyty plazmatyczne, które po pewnych transformacjach zaczynają produkować ochronne białka globularne, czyli gammaglobuliny (immunoglobuliny lub przeciwciała).
Patogeneza chłoniaka limfoplazmocytowego/makroglobulinemii Waldenströma obejmuje hiperproliferację komórek B, nadmiar klonu komórek plazmatycznych limfocytów i nadmierną produkcję immunoglobuliny M (IgM), zwanej również monoklonalną immunoglobuliną lub białkiem M, we krwi. Jest to główne przeciwciało o dużej masie cząsteczkowej i strukturze pentamerycznej, wytwarzane podczas początkowego ataku na określone antygeny bakteryjne lub wirusowe. [ 5 ]
Prawie wszystkie objawy tej choroby wiążą się z przejawami aktywności białka M, które może zaburzać właściwości reologiczne krwi, zwiększać jej lepkość, przenikać do tkanek limfoidalnych i mieloidalnych szpiku kostnego, gromadzić się w obwodowych tkankach limfoidalnych (z powstawaniem powoli rosnących nowotworów zdolnych wywierać nacisk na otaczające narządy, włókna nerwowe lub naczynia krwionośne).
Mimo że przewlekła białaczka limfocytowa, makroglobulinemia Waldenströma lub chłoniak limfoplazmocytowy i szpiczak mnogi to odrębne choroby, wszystkie one wiążą się ze wzmożoną proliferacją limfocytów B.
Objawy chłoniak limfoplazmocytowy
Pierwsze objawy choroby są niespecyficzne i mogą obejmować osłabienie i zwiększone zmęczenie (z powodu rozwoju niedokrwistości normochromowej), utratę masy ciała, duszność, nocne pocenie się i nawracającą gorączkę podgorączkową.
Ponadto w początkowym stadium choroby dochodzi do zaburzeń czucia dłoni i stóp, pojawia się neuropatia obwodowa (drętwienie lub mrowienie stóp i nóg), pojawiają się drobne ogniskowe krwawienia naczyń włosowatych skóry (plamica), a także pokrzywka z zimna (wskutek powstawania i agregacji nieprawidłowych białek krioglobuliny w surowicy krwi).
Objawy związane z zespołem hiperlepkości obejmują bóle głowy i zawroty głowy, uszkodzenie siatkówki i utratę wzroku, szumy uszne i utratę słuchu, skurcze, bóle mięśni, wysokie ciśnienie krwi, samoistne krwawienia z nosa i krwawienie dziąseł. U kobiet może wystąpić krwawienie z macicy.
Zaobserwowano również: powiększone węzły chłonne (limfadenopatia); powiększoną śledzionę (splenomegalia); niewydolność serca z kardialgią i zaburzeniami rytmu serca. Chociaż naciek trzewny jest rzadki, żołądek i jelita mogą być dotknięte, z rozwojem biegunki (często z tłustymi stolcami). [ 6 ], [ 7 ]
Formularze
Klasyfikacja nowotworów tkanek krwiotwórczych i limfatycznych Światowej Organizacji Zdrowia z 2017 r. ustala cztery kryteria diagnostyczne makroglobulinemii Waldenströma, w tym:
- Obecność monoklonalnej gammapatii IgM
- Naciekanie szpiku kostnego przez małe limfocyty wykazujące różnicowanie plazmocytoidalne lub różnicowanie komórek plazmatycznych
- Naciek szpiku kostnego ze strukturą międzybeleczkową
- Immunofenotyp zgodny z makroglobulinemią Waldenströma, obejmujący powierzchniowe IgM+, CD19+, CD20+, CD22+, CD25+, CD27+, FMC7+, zmienne CD5, CD10-, CD23-, CD103- i CD108-
Komplikacje i konsekwencje
U chorych na chłoniaka limfoplazmocytowego rozwijają się powikłania i konsekwencje w postaci:
- obniżona odporność;
- niewydolność szpiku kostnego z zaburzeniem jego funkcji krwiotwórczych i rozwojem anemii;
- niedobór takich elementów krwi jak erytrocyty, leukocyty, płytki krwi;
- uszkodzenia przewodu pokarmowego z przewlekłą biegunką i zaburzeniami wchłaniania jelitowego (zespół złego wchłaniania);
- zapalenie ścian naczyń krwionośnych (złożone zapalenie naczyń o podłożu immunologicznym);
- zwiększona kruchość kości (osteoporoza);
- wady wzroku i słuchu;
- wtórna amyloidoza narządów wewnętrznych;
- progresja do paraproteinemicznej hemoblastozy w postaci szpiczaka mnogiego;
- przekształcenie się w wysoce złośliwy typ chłoniaka – chłoniaka rozlanego z dużych komórek B.
Diagnostyka chłoniak limfoplazmocytowy
Rozpoznanie chłoniaka limfoplazmocytowego/makroglobulinemii Waldenströma jest zazwyczaj trudne ze względu na brak specyficznych zmian morfologicznych, immunofenotypowych lub chromosomalnych. Ten niedobór sprawia, że różnicowanie tej choroby od innych chłoniaków małych komórek B jest kwestią wykluczenia.[ 8 ]
Aby rozpoznać chłoniaka limfoplazmocytowego, oprócz oceny istniejących objawów konieczne jest wykonanie ogólnego i biochemicznego badania krwi, koagulogramu, immunoelektroforezy białek krwi z oznaczeniem poziomu immunoglobuliny M we krwi oraz ogólnego badania moczu. [ 9 ]
Wymagana jest biopsja szpiku kostnego, w ramach której wykonuje się nakłucie szpiku kostnego.
Wykonuje się diagnostykę instrumentalną: USG węzłów chłonnych i śledziony, RTG kości, tomografię komputerową klatki piersiowej i jamy brzusznej, oftalmoskopię.
Diagnostyka różnicowa
Chłoniak limfoplazmocytowy jest uważany za rozpoznanie z wykluczenia, dlatego diagnostykę różnicową przeprowadza się z przewlekłą białaczką limfocytową B-komórkową, szpiczakiem mnogim, chłoniakiem grudkowym, różnymi podtypami chłoniaka nieziarniczego, plazmocytomą, reaktywną plazmocytozą, naczyniowo-grudkową hiperplazją limfoidalną (chorobą Castlemana) itp.
Z kim się skontaktować?
Leczenie chłoniak limfoplazmocytowy
Należy pamiętać, że makroglobulinemia Waldenströma, czyli chłoniak limfoplazmocytowy, może przez wiele lat przebiegać bezobjawowo, a rozpoznanie można postawić na podstawie wzrostu poziomu białka M we krwi.
W przypadku braku objawów należy prowadzić aktywną obserwację za pomocą regularnych badań i testów.
Na podstawie występujących objawów oraz wyników badań laboratoryjnych podejmowana jest decyzja o rozpoczęciu terapii, która zależy od wielu czynników (np. wieku, zaawansowania choroby itp.).
Zgodnie z protokołem, początkowe leczenie pacjentów z tym typem chłoniaka obejmuje zazwyczaj połączenie radioterapii i chemioterapii z podawaniem cytostatyków, takich jak cyklofosfamid, doksorubicyna, winkrystyna, a także kortykosteroidów - metprednizolonu lub deksametazonu (Dexasone).
Udowodniono skuteczność chemioterapii lekami z grupy przeciwciał monoklonalnych, w szczególności rytuksymabu. [10 ]
W przypadku choroby uogólnionej Rituximab stosuje się w połączeniu z przeciwnowotworowymi analogami nukleozydów (Pentostatyna, Kladrybina). W przypadku choroby o powolnym postępie, z niskim poziomem monoklonalnej immunoglobuliny M, oprócz Rituximabu stosuje się cytostatyk Chlorambucil (Leukeran). [ 11 ]
Aby zmniejszyć lepkość krwi i ustabilizować poziom jej składników, stosuje się leczniczą hemaferezę.
Gdy poziom przeciwciał we krwi jest krytycznie niski, stosuje się terapię zastępczą immunoglobulinami, aby zapobiec nawracającym zakażeniom.
Jak zauważają onkohematolodzy, mimo że leczenie może doprowadzić do remisji choroby, u większości pacjentów dochodzi do nawrotu. Jeśli nastąpi on wcześniej niż po 24 miesiącach, można zastosować lek przeciwnowotworowy, np. Ibrutynib (w postaci tabletek). W przypadku późniejszych nawrotów leczenie prowadzi się według schematu pierwotnego. [ 12 ], [ 13 ], [ 14 ]
Zapobieganie
Eksperci określają rokowanie co do wyniku chłoniaka limfoplazmocytowego według międzynarodowego systemu prognostycznego, oceniającego główne parametry: wiek pacjenta oraz stężenie hemoglobiny, płytek krwi, beta-2-mikroglobuliny i monoklonalnej immunoglobuliny w surowicy. [ 15 ], [ 16 ]
Średni wskaźnik przeżycia w przypadku tej diagnozy wynosi około pięciu lat, ale prawie 40% pacjentów przeżywa dziesięć lat lub dłużej.