
Cała zawartość iLive jest sprawdzana medycznie lub sprawdzana pod względem faktycznym, aby zapewnić jak największą dokładność faktyczną.
Mamy ścisłe wytyczne dotyczące pozyskiwania i tylko linki do renomowanych serwisów medialnych, akademickich instytucji badawczych i, o ile to możliwe, recenzowanych badań medycznych. Zauważ, że liczby w nawiasach ([1], [2] itd.) Są linkami do tych badań, które można kliknąć.
Jeśli uważasz, że któraś z naszych treści jest niedokładna, nieaktualna lub w inny sposób wątpliwa, wybierz ją i naciśnij Ctrl + Enter.
Lissencefalia mózgu
Ekspert medyczny artykułu

Ostatnia recenzja: 12.07.2025
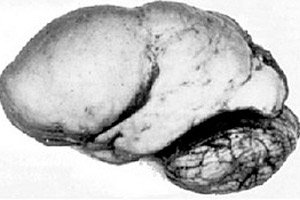
Wśród patologii organicznych mózgu wyróżnia się wrodzona anomalia rozwoju mózgu, jaką jest lissencephalia, której istota polega na niemal gładkiej powierzchni kory jej półkul - z niedostateczną liczbą zwojów i bruzd. [ 1 ]
W przypadku całkowitego braku zakrętów definiuje się agyrię, a obecność kilku szerokich płaskich zakrętów nazywa się pachygyrią. Te defekty, podobnie jak niektóre inne deformacje redukcyjne mózgu, mają kod Q04.3 w ICD-10.
Epidemiologia
Według statystyk dotyczących chorób rzadkich, na 100 tys. noworodków przypada 1-1,2 przypadków lissencephalii. [ 2 ], [ 3 ]
Według niektórych danych u dzieci z zespołem Millera-Diekera obserwuje się nawet 25-30% przypadków klasycznej lisencefalii, przy czym u blisko 85% pacjentów wykrywa się mutacje punktowe i delecje genów LIS1 i DCX. [ 4 ]
Badania genetyczne 17 genów związanych z lissencephalią wykazały, że mutacja lub usunięcie genu LIS1 odpowiada za 40% przypadków pacjentów, a 23% jest związanych z mutacją genu DCX, następnie TUBA1A (5%) i DYNC1H1 (3%).[ 5 ]
Przyczyny lissencefalia
Wszystkie znane przyczyny powstawania kory mózgowej (cortex cerebri) niemal lub całkowicie pozbawionej zakrętów i bruzd, które zwiększają „obszar roboczy” mózgu ludzkiego i zapewniają „produktywność” ośrodkowego układu nerwowego, wiążą się z zaburzeniami w jej rozwoju okołoporodowym. Mianowicie u płodu rozwija się lissencephalia. [ 6 ]
Niepowodzenie w formowaniu się warstw kory mózgowej u płodu w przypadku lisencefalii jest wynikiem nieprawidłowej migracji neuronów, które ją tworzą, lub przedwczesnego zatrzymania tego procesu.
Proces ten, niezbędny dla histogenezy kory mózgowej, zachodzi w kilku etapach od 7 do 18 tygodnia ciąży. A biorąc pod uwagę jego zwiększoną wrażliwość na mutacje genetyczne, a także różne negatywne wpływy fizyczne, chemiczne i biologiczne, każde odstępstwo od normy może prowadzić do nieprawidłowej lokalizacji neuronów z możliwym utworzeniem pogrubionej warstwy istoty szarej kory mózgowej bez charakterystycznej struktury. [ 7 ]
W niektórych przypadkach lisencefalia u dzieci jest związana z zespołem Millera-Diekera, Walkera-Warburga lub Normana-Robertsa.
Przeczytaj także – Wady rozwojowe mózgu
Czynniki ryzyka
Czynnikami ryzyka urodzenia dziecka z tak ciężką wadą, oprócz mutacji w niektórych genach, są: niedotlenienie płodu (niedotlenienie), niedostateczne ukrwienie mózgu (hipoperfuzja), ostry udar mózgu w postaci udaru okołoporodowego, patologie łożyska, zakażenia wirusowe u kobiety ciężarnej (w tym zespół TORCH); [ 8 ] problemy z ogólnym metabolizmem i funkcją tarczycy; palenie tytoniu, spożywanie alkoholu, substancji psychotropowych i narkotycznych; stosowanie szeregu leków; zwiększone stężenie promieniowania. [ 9 ]
Patogeneza
Nie wszystkie przypadki lissencephalii mają patogenezę spowodowaną nieprawidłowościami chromosomowymi i mutacjami genów. Ale znane są pewne geny, które kodują białka odgrywające ważną rolę w prawidłowym ruchu neuroblastów i neuronów wzdłuż komórek glejowych promienistych - w celu utworzenia kory mózgowej. A mutacje tych genów prowadzą do tej patologii. [ 10 ]
W szczególności są to sporadyczne mutacje (bez dziedziczności) genu LIS1 na chromosomie 17, który reguluje cytoplazmatyczne białko motoryczne mikrotubul dyneiny, a także genu DCX na chromosomie X, który koduje białko doublecortin (lissencephalin-X). [ 11 ]. W pierwszym przypadku specjaliści określają klasyczną lissencephalię (typ I), w drugim – sprzężoną z chromosomem X. [ 12 ]
Gdy gen FLN1, kodujący fosfoproteinę filaminę 1, zostanie usunięty, proces ukierunkowanej migracji neuronów może w ogóle się nie rozpocząć, co doprowadzi do całkowitego braku splotów (agyrii). [ 13 ]
Zidentyfikowano mutacje w genie CDK5, który koduje enzym kinazę – katalizator metabolizmu wewnątrzkomórkowego, regulujący cykl komórkowy w neuronach ośrodkowego układu nerwowego i zapewniający ich prawidłową migrację podczas prenatalnego kształtowania się struktur mózgu.
Nieprawidłowe zmiany w genie RELN na chromosomie 7, które powodują uszkodzenia zakrętów korowych w zespole Normana-Robertsa, skutkują niedoborem pozakomórkowego glikoproteiny reeliny, która jest wymagana do regulacji migracji i pozycjonowania komórek macierzystych układu nerwowego podczas rozwoju kory mózgowej.[ 14 ], [ 15 ], [ 16 ]
Gen ARX koduje białko homeoboksowe niearystalens, czynnik transkrypcyjny odgrywający ważną rolę w przednim mózgu i innych tkankach.[ 17 ] U dzieci z mutacją ARX występują również inne objawy, takie jak brak części mózgu (agenezja ciała modzelowatego), nieprawidłowe narządy płciowe i ciężka padaczka.[ 18 ],[ 19 ]
Z lissencephalią powiązano kilka genów. Należą do nich geny VLDLR, ACTB, ACTG1, TUBG1, KIF5C, KIF2A i CDK5.[ 20 ]
Cytomegalovirus (CMV) został powiązany z rozwojem lissencephalii z powodu zmniejszonego dopływu krwi do mózgu płodu. Ciężkość zakażenia CMV zależy od wieku ciążowego. Wczesne zakażenie jest bardziej prawdopodobne, aby spowodować lissencephalię, ponieważ migracja neuronów występuje wcześnie w ciąży.[ 21 ]
Ponadto mechanizm powstawania tej anomalii obejmuje niepełne lub późniejsze ustanie migracji neuronów z okołokomorowej strefy generatywnej do kory mózgowej. I w takich przypadkach rozwija się albo niepełna lissencephalia, albo pachygyria, w której tworzy się kilka szerokich bruzd i zakrętów (ale większość z nich jest nieobecna).
Objawy lissencefalia
Pierwsze objawy tej patologii (przy braku wcześniej wymienionych zespołów) mogą nie pojawić się bezpośrednio po urodzeniu, ale po półtora do dwóch miesięcy. I najczęściej obserwuje się następujące objawy kliniczne lissencephalii:
- hipotonia mięśniowa, często połączona z porażeniem spastycznym;
- drgawki i napady uogólnione toniczno-kloniczne (w postaci opistotonusu);
- ciężkie upośledzenie umysłowe i zahamowanie wzrostu;
- upośledzenie funkcji neurologicznych i motorycznych.
Problemy z połykaniem powodują trudności w karmieniu dziecka. [ 22 ]
Wysoki stopień upośledzenia neuromotorycznego często objawia się tetraplegią – paraliżem wszystkich kończyn. Możliwe są deformacje dłoni, palców rąk lub stóp.
W zespole Normana-Robertsa z lisencefalią typu I obserwuje się anomalie czaszkowo-twarzowe: ciężką małogłowie, niskie nachylenie czoła i wystający szeroki grzbiet nosa, szeroko rozstawione oczy (hiperterloryzm), niedorozwój szczęk (mikrognacja). [ 23 ]
Zespół Millera-Diekera może również charakteryzować się nieprawidłowo małą głową z szerokim, wysokim czołem i krótkim nosem, zagłębieniami skroniowymi (zagłębieniami dwuskroniowymi) oraz nisko osadzonymi, zdeformowanymi uszami.
Ciężki zespół lisencefalii charakteryzuje się małogłowiem, czyli zmniejszeniem wielkości gałek ocznych (mikroftalmia), w połączeniu z dysplazją siatkówki, wodogłowiem obturacyjnym oraz brakiem lub niedorozwojem ciała modzelowatego.
Komplikacje i konsekwencje
Wśród powikłań tej anomalii specjaliści wymieniają zaburzenie funkcji połykania (dysfagię) i refluks żołądkowo-przełykowy; padaczkę oporną na leczenie (niekontrolowaną); częste infekcje górnych dróg oddechowych; zapalenie płuc (w tym przewlekłe zachłystowe).
U niemowląt z lisencefalią mogą występować wrodzone organiczne problemy kardiologiczne w postaci ubytku przegrody międzyprzedsionkowej lub złożonej wady serca z sinicą (tetralogia Fallota). [ 24 ]
Konsekwencją postnatalnego zahamowania wzrostu jest w większości przypadków śmierć w ciągu 24 miesięcy od urodzenia.
Diagnostyka lissencefalia
Diagnozę stawia się na podstawie badania fizykalnego dziecka, analizy historii medycznej rodziców oraz przebiegu ciąży i porodu.
W czasie ciąży może być konieczne wykonanie badania pozakomórkowego DNA płodu, amniopunkcji lub biopsji kosmówki. [ 25 ] Aby uzyskać więcej informacji, zobacz – Diagnostyka prenatalna chorób wrodzonych
Diagnostyka instrumentalna służy do obrazowania struktur mózgowych i oceny ich funkcji:
- tomografia komputerowa mózgu;
- obrazowanie mózgu metodą rezonansu magnetycznego (MRI);
- elektroencefalogram (EEG). [ 26 ]
W czasie ciąży, w badaniu USG płodu po 20-21 tygodniu ciąży, można podejrzewać lisencefalię, jeśli u płodu nie ma bruzdy ciemieniowo-potylicznej i bruzdy ostrogowej, a także występuje anomalia szczeliny Sylwiusza mózgu.
Diagnostyka różnicowa
Przeprowadza się diagnostykę różnicową z innymi zespołami wrodzonych wad mózgu.
Istnieje ponad 20 typów lissencephalii, z których większość mieści się w 2 głównych kategoriach: klasycznej lissencephalii (typ 1) i brukowanej lissencephalii (typ 2). Każda kategoria ma podobne objawy kliniczne, ale różne mutacje genetyczne.[ 27 ]
Badanie mózgu w przypadku lissencephalii typu I pokazuje korę mózgową z czterema warstwami zamiast sześciu widocznych u zdrowych pacjentów, podczas gdy w przypadku lissencephalii typu 2 kora mózgowa jest zdezorganizowana i wydaje się grudkowata lub guzkowata z powodu całkowitego przemieszczenia kory mózgowej przez skupiska neuronów korowych rozdzielonych tkanką glejo-mezenchymalną. Pacjenci mieli również nieprawidłowości mięśni i oczu.
- Klasyczna lissencephalia (typ 1):
- LIS1: Izolowana lissencephalia i zespół Millera-Diekera (lissencephalia związana z dysmorfią twarzy). [ 28 ]
- LISX1: Mutacja genu DCX. W porównaniu do lissencephalii spowodowanej mutacjami LIS1, DCX wykazuje sześciowarstwową korę zamiast czterowarstwowej.
- Izolowana lisencefalia bez innych znanych wad genetycznych
- Lisencefalia typu brukowanego (typ 2):
- Zespół Walkera-Warburga
- Syndrom Fukuyamy
- Choroba mięśni, oczu i mózgu
- Pozostałych typów nie można umieścić w żadnej z dwóch powyższych grup:
- LIS2: Zespół Normana-Robertsa, podobny do lisencefalii typu I lub zespołu Millera-Diekera, lecz bez delecji chromosomu 17.
- LIS3
- LISX2
Mikrolissencephalia: Jest to połączenie braku prawidłowego fałdowania kory mózgowej i nienormalnie małej głowy. Dzieci z regularną lissencephalią mają głowy normalnej wielkości przy urodzeniu. U dzieci z małą głową przy urodzeniu zwykle diagnozuje się mikrolissencephalię.
Ważne jest również rozróżnienie lisencefalii i polimikrogyrii, które są różnymi wadami rozwojowymi mózgu.
Z kim się skontaktować?
Leczenie lissencefalia
Lissencephalia jest nieuleczalną wadą organiczną, dlatego możliwe jest jedynie leczenie podtrzymujące i objawowe. [ 29 ]
Przede wszystkim jest to stosowanie leków przeciwdrgawkowych i przeciwpadaczkowych, a także założenie rurki gastrostomijnej do żołądka (jeśli dziecko nie jest w stanie samodzielnie połykać). Przydatny jest masaż.
W przypadkach ciężkiego wodogłowia usuwa się płyn mózgowo-rdzeniowy.
Zapobieganie
Eksperci zalecają, aby przyszli rodzice skorzystali z poradnictwa genetycznego, a kobiety w ciąży, aby odpowiednio wcześnie zgłaszały się do położników i ginekologów oraz poddawały się wszystkim zaplanowanym badaniom.
Prognoza
U dzieci z lissencephalią rokowanie zależy od jej stopnia, ale najczęściej rozwój umysłowy dziecka nie przekracza poziomu czterech-pięciu miesięcy. A wszystkie dzieci z tą diagnozą cierpią na ciężkie zaburzenia psychoruchowe i trudną do leczenia padaczkę. [ 30 ]
Według NINDS (Amerykańskiego Narodowego Instytutu Zaburzeń Neurologicznych i Udaru Mózgu) maksymalna długość życia osób chorych na lissencephalię wynosi około 10 lat.