
Cała zawartość iLive jest sprawdzana medycznie lub sprawdzana pod względem faktycznym, aby zapewnić jak największą dokładność faktyczną.
Mamy ścisłe wytyczne dotyczące pozyskiwania i tylko linki do renomowanych serwisów medialnych, akademickich instytucji badawczych i, o ile to możliwe, recenzowanych badań medycznych. Zauważ, że liczby w nawiasach ([1], [2] itd.) Są linkami do tych badań, które można kliknąć.
Jeśli uważasz, że któraś z naszych treści jest niedokładna, nieaktualna lub w inny sposób wątpliwa, wybierz ją i naciśnij Ctrl + Enter.
Keratodermia: przyczyny, objawy, diagnoza, leczenie
Ekspert medyczny artykułu

Ostatnia recenzja: 07.07.2025
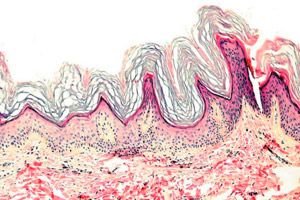
Keratodermia to grupa chorób skóry charakteryzująca się zaburzeniem procesu rogowacenia - nadmiernym tworzeniem się naskórka, głównie na dłoniach i podeszwach stóp.
Przyczyny i patogeneza choroby nie zostały do końca wyjaśnione. Badania wykazały, że keratodermy są spowodowane mutacjami w genach kodujących keratynę 6, 9, 16. Niedobór witaminy A, zaburzenia hormonalne, przede wszystkim gruczołów płciowych, infekcje bakteryjne i wirusowe mają duże znaczenie w patogenezie. Są jednym z objawów chorób dziedzicznych i nowotworów narządów wewnętrznych (keratodermy parapsoriatyczne).
Objawy. Rozróżnia się rozproszoną (keratodermia Unny-Tosta, keratodermia Meledy, keratodermia Papillon-Lefevre'a, keratodermia okaleczająca i zespoły, w których jednym z głównych objawów jest rozproszona keratodermia) i ogniskową (rozsiana plamista keratodermia Fischera-Buschkego, akrokeratoelastoidoza Kostiego, ograniczona keratodermia Bruhauera-Franzesthestiego, linijna keratodermia Fuchsa itp.) keratodermia.
Winy-Tost keratodermia (synonimy: wrodzona rybia łuska dłoni i podeszew, zespół Winy-Tost) jest dziedziczona w sposób autosomalny dominujący. Występuje rozlane nadmierne rogowacenie skóry dłoni i podeszew (czasem tylko podeszew), które rozwija się w pierwszych dwóch latach życia. Proces patologiczny skóry rozpoczyna się od lekkiego zgrubienia skóry dłoni i podeszew w postaci pasma rumienia o sinoczerwonym kolorze na granicy ze zdrową skórą. Z czasem na ich powierzchni pojawiają się gładkie, żółtawe, rogowe warstwy. Zmiana rzadko rozprzestrzenia się na grzbiet nadgarstków lub palców. U niektórych pacjentów mogą tworzyć się powierzchowne lub głębokie pęknięcia i obserwuje się miejscową nadpotliwość. U pacjenta obserwowanego przez autora na Winy-Tost chorował wujek ze strony matki, brat i syn.
Opisano przypadki uszkodzeń paznokci (zgrubienia), zębów i włosów w keratodermii Winy-Tosta w połączeniu z różnymi anomaliami szkieletowymi i patologiami narządów wewnętrznych, układu nerwowego i hormonalnego.
Histopatologia. Badanie histologiczne ujawnia wyraźną hiperkeratozę, ziarniniak, akantozę i małe nacieki zapalne w górnej warstwie skóry właściwej. Diagnostyka różnicowa. Chorobę należy odróżnić od innych typów keratodermy.
Meleda keratoderma (synonimy: choroba Meleda, wrodzony postępujący akrokeratoma, transgradientowe rogowacenie dłoni i stóp Siemensa, dziedziczne postępujące rogowacenie dłoni i stóp Kogoya) dziedziczona jest w sposób autosomalny recesywny. Ta postać keratodermy charakteryzuje się grubymi, żółtobrązowymi warstwami rogowymi z głębokimi pęknięciami. Fioletowo-purpurowa obwódka o szerokości kilku milimetrów jest widoczna wzdłuż krawędzi zmiany. Proces ten zwykle rozprzestrzenia się na tylną część dłoni i stóp, przedramiona i piszczele. Większość pacjentów doświadcza miejscowej nadpotliwości. W związku z tym powierzchnia dłoni i podeszew stóp staje się lekko wilgotna i pokryta czarnymi kropkami (przewody gruczołów potowych).
Choroba może rozwinąć się już w wieku 15-20 lat. Paznokcie stają się grube i zdeformowane.
Histopatologia. Badanie histologiczne ujawnia hiperkeratozę, czasami akantozę i przewlekły naciek zapalny w brodawkowatej skórze właściwej.
Diagnostyka różnicowa. Melela keratoderma musi być odróżniona od Unna-Tost keratoderma.
Keratodermia Papillon-Lefevre'a (synonim: nadmierne rogowacenie dłoni i stóp z zapaleniem przyzębia) dziedziczona jest w sposób autosomalny recesywny.
Choroba ujawnia się w 2-3 roku życia. Obraz kliniczny choroby jest podobny do choroby Meleli. Ponadto charakterystyczne są zmiany w uzębieniu (nieprawidłowości w wyrzynaniu się zębów mlecznych i stałych z rozwojem próchnicy, zapalenie dziąseł, szybko postępująca paradontoza z przedwczesną utratą zębów).
Histopatologia. Badanie histologiczne ujawnia pogrubienie wszystkich warstw naskórka, szczególnie warstwy rogowej, oraz nieznaczne skupiska komórek limfocytów i histiocytów w skórze właściwej.
Diagnostyka różnicowa. Chorobę należy odróżnić od innych keratoderm. Ważną cechą odróżniającą jest charakterystyczna patologia zębów, której nie stwierdza się w innych formach dziedzicznych rozproszonych keratoderm.
Keratoderma mutilans (synonimy: zespół Fonwinkla, dziedziczny okaleczający keratoma) jest typem rozlanego keratodermy dziedziczonym w sposób autosomalny dominujący. Rozwija się w 2. roku życia i charakteryzuje się rozproszonymi rogowymi złogami na skórze dłoni i stóp z nadmierną potliwością. Z czasem na palcach tworzą się bruzdy przypominające sznury, co prowadzi do przykurczów i samoistnej amputacji palców. Rogowacenie mieszkowe jest wyrażone na grzbietach dłoni, a także w okolicy stawów łokciowych i kolanowych. Płytki paznokciowe są zmienione (często jak szkła zegarkowe). Opisano przypadki hipogonadyzmu, łysienia rubinowego, utraty słuchu, pachyonychii.
Histopatologia. Badanie histologiczne ujawnia ciężką hiperkeratozę, ziarniniak, akantozę i małe nacieki zapalne w skórze właściwej, składające się z limfocytów i histiocytów.
Diagnostyka różnicowa. Różnicując keratodermę okaleczającą od innych postaci rozlanej keratodermy, należy przede wszystkim wziąć pod uwagę efekt okaleczenia, który nie jest typowy dla innych postaci. Przeprowadzając diagnostykę różnicową wszystkich postaci rozlanej keratodermy, należy pamiętać, że może ona być jednym z głównych objawów wielu zespołów dziedzicznych.
Leczenie. Neotigazon jest wskazany w ogólnej terapii keratodermy. Dawka leku zależy od nasilenia procesu i wynosi 0,3-1 mg/kg masy ciała pacjenta. W przypadku braku neotigazonu zaleca się witaminę A w dawce od 100 do 300 000 mg na dobę przez długi czas. Terapia zewnętrzna polega na stosowaniu maści z aromatycznymi retinoidami, środkami keratolitycznymi i steroidowymi.
 [
[ Co Cię dręczy?
Co trzeba zbadać?
Jak zbadać?